Su quali basi e dopo quali”rigorosi” controlli, viene rilasciata la licenza prima di immetterli sul mercato per la commercializzazione ?
Aggiornamento fonti novembre 2015 ( a fine articolo)
Oltre un miliardo di dosi di vaccini prodotti in tutto il mondo ogni anno, sono indicati e vengono somministrati attraverso la profilassi vaccinale, a neonati sani, bambini e adulti.
Pertanto, è fondamentale che i vaccini dimostrino di essere sicuri ed efficaci.
L’organo FDA richiede per questo motivo un rigoroso programma di sviluppo e di controllo in laboratorio, così come viene fatto negli studi su animali e clinici, per determinare la loro sicurezza ed efficacia. Scienziati e clinici dell’ FDA dovrebbero quindi valutare attentamente il tutto per poi rilasciare la licenza dando il via alla commercializzazione negli Stati Uniti.
Prima di rilasciare la licenza,l’ FDA prende tutti gli ingredienti di un vaccino in considerazione, compresi quelli attivi. Dopo che la stessa FDA approva un vaccino,successivamente passa al controllo della sua sicurezza.
Perché in alcuni vaccini è presente l’alluminio ?
I Sali di alluminio sono incorporati in alcune formulazioni, come coadiuvante.
Un adiuvante è una sostanza aggiunta ad alcuni vaccini per “migliorare” la risposta immunitaria dei soggetti vaccinati. I sali di alluminio in alcuni vaccini autorizzati sono:
idrossido di alluminio, fosfato di alluminio, allume (solfato di alluminio di potassio), o sali di alluminio misti.
Per esempio: sali di alluminio sono utilizzati nei vaccini DTaP, il vaccino pneumococcico coniugato, e vaccini per l’epatite B.
***CONTINUANDO LA “FARSA” DELLA “VERITA” DA PARTE DI QUESTI COMPETENTI ORGANI, SI LEGGE QUANTO SEGUE:
“L’Alluminio contenuto nei vaccini ha un profilo di sicurezza dimostrato da oltre sei decenni di utilizzo. Da segnalare, la fonte più comune di esposizione all’alluminio è da mangiare cibo o acqua potabile”
***Ricordiamo :
ANCORA OGGI SI NEGA la presenza dell’Alluminio nei vaccini e di altri elementi quali Mercurio etc..
QUESTI ELEMENTI sono tossici,in quanto tali arrivano nei nostri corpi in maniera “INNATURALE” (poichè intramuscolo) forzando lo stesso a superare o a scavalcare addirittura -cosa pericolosissima- quegli step fondamentali per combattere e debellare nella giusta maniera i virus. L’esempio dell’aria o dell’acqua non è paragonabile ad una iniezione, questo è bene dirlo.
TAVOLA CONTENENTE TUTTI GLI ELEMENTI
Vaccine Excipient & Media Summary Excipients Included in U.S. Vaccines, by Vaccine
Ci sono altri coadiuvanti utilizzati nei vaccini approvati dalla FDA?
Sì. Cervarix, un vaccino per prevenire il cancro del collo dell’utero causato da diversi tipi di papillomavirus umano (16 e 18), comprende AS04 nella sua formulazione.
L’AS04 è una combinazione di idrossido di alluminio e monofosforil lipide A (MPL).
L’MPL è una sostanza simile al grasso purificato.
Inoltre, il vaccino per la prevenzione dell’ H5N1, comunemente indicata come “influenza aviaria,” contiene l’adiuvante AS03, una emulsione “olio-in-acqua”. L’adiuvante AS03 è costituito dai composti oleosi, D, L-alfa-tocoferolo (vitamina E), squalene, e il polisorbato 80, il quale serve a mantenere insieme tutti gli elementi , impedendo loro di separarsi; poi troviamo acqua contenente piccole quantità i sali .
Come l’ FDA valuta la sicurezza e l’efficacia?
Quando si valuta l’efficacia e la sicurezza di un vaccino, l’ FDA considera il comportamento e le reazioni,la nocività degli adiuvanti come componente di esso; questi non sono concessi in licenza separatamente.
Perché la presenza di antibiotici in alcuni vaccini?
Alcuni antibiotici possono essere utilizzati in determinate produzioni per “prevenire la contaminazione batterica durante la produzione“.
Il risultato? Piccole quantità di antibiotici possono essere presenti in alcuni vaccini.
Ricordiamo come alcuni antibiotici possano causare gravi reazioni allergiche in molti bambini allergici a loro volta (orticaria, gonfiore nella parte posteriore della gola, e la pressione sanguigna bassa); i genitori sono preoccupati perciò della dannosità di tali antibiotici presenti.
Tuttavia,dichiarano che gli antibiotici più suscettibili, causa delle gravi reazioni allergiche (ad esempio, penicilline, cefalosporine e sulfamidici) non sono usati nella produzione di vaccini, e quindi non sono contenuti nei vaccini.
Esempi di antibiotici utilizzati durante la fabbricazione di vaccini includono neomicina, polimixina B, streptomicina e gentamicina. Alcuni antibiotici utilizzati nella produzione di vaccini sono presenti in quantità molto piccole.
Un esempio? I vaccini dell’influenza con virus “inattivato”.
Essi sono utilizzati per ridurre la crescita batterica nelle uova, durante le fasi di lavorazione, perché queste, non sono prodotti sterili. Gli antibiotici che vengono utilizzati a dosi molto piccole,non sono stati chiaramente associati a gravi reazioni allergiche.
***E’ BENE RICORDARE SEMPRE AI DOTTORI DI FARE CONSENSO INFORMATO E DI CHIEDERE EVENTUALI PREDISPOSIZIONI IN FAMIGLIA. NOI SIAMO IL NOSTRO CORREDO GENETICO.A CONTRASTARE QUESTE DICHIARAZIONI LEGGIAMO DI SEGUITO L’INEFFICACIA E L’ALLARME PER LE INFEZIONI***
L’inefficacia degli antibiotici. Un antibiotico su due è inefficace: è allarme per le infezioni. L’inefficacia degli antibiotici; un antibiotico su due non funziona. Essere resistenti ai farmaci è un fenomeno che dilaga ed’è abbastanza preoccupante: un antibiotico su due, infatti, non funziona.
Perché la presenza di formaldeide in alcuni vaccini?
La Formaldeide ha una lunga storia dell’ uso sicuro nella fabbricazione di alcuni vaccini virali e batterici. Viene utilizzata per inattivare virus in modo che essi non causino la malattia (ad esempio, per virus della polio, usato per comporre il vaccino) e decontaminare tossine batteriche, come la tossina usata per il vaccino contro la difterite.
La formaldeide è diluita durante il processo di fabbricazione, ma i quantitativi residui di formaldeide possono essere trovati in alcuni vaccini attuali.
“La quantità di formaldeide presente in alcuni vaccini è così piccola rispetto alla concentrazione che si verifica naturalmente nel corpo che non pone problemi di sicurezza”.
La formaldeide è anche prodotta naturalmente nel corpo per produrre energia e creando il materiale di base necessario per gli importanti processi vitali. Ciò include gli amminoacidi, che sono i mattoni delle proteine di cui il corpo ha bisogno.
La formaldeide è presente anche nell’ambiente in diversi modi. E ‘utilizzata nei materiali da costruzione, come conservante nei laboratori e per la produzione di molti prodotti per la casa.
L’eccessiva esposizione alla formaldeide può causare il cancro, ma le ultime ricerche hanno dimostrato che il rischio maggiore è portato dall’aria quando la formaldeide viene respirata (si verifica spesso nelle persone che abitualmente utilizzano la formaldeide nel loro lavoro).
Non vi è alcuna prova che collega il cancro all’esposizione infrequente piccole quantità di formaldeide tramite iniezione come avviene con i vaccini.
***ALLEGHIAMO DI SEGUITO UN ASTRATTO CON FONTE CHE SI CONTRAPPONE CON LE DICHIARAZIONI ERRATE DELL’ORGANO FDA***
LA SICUREZZA DELLA FORMALDEIDE E’ CERTIFICATA: CANCEROGENA!
Formaldeide cancerogena
Inserito il 1 aprile 2015 in News ANMA
Il 24 marzo è stato pubblicato sulla Gazzetta Ufficiale dell’Unione Europea il Regolamento (UE) 2015/491 del 23 marzo 2015
Ultim’ora: il 24 marzo è stato pubblicato sulla Gazzetta Ufficiale dell’Unione Europea il Regolamento (UE) 2015/491 del 23 marzo 2015 che, facendo seguito a notizie già diffuse in forma ufficiosa nelle scorse settimane, conferma lo spostamento del termine del periodo transitorio per l’applicazione alla formaldeide della nuova classificazione come cancerogeno di categoria 1B al 1 gennaio 2016.
La data precedentemente prevista era il 1 aprile 2015, lo spostamento si è reso necessario in quanto il periodo transitorio previsto tra la pubblicazione del regolamento 605/2014 che ha sancito il cambio di classificazione e la data prevista per l’entrata in vigore è stato ritenuto troppo breve rispetto ad altri casi analoghi.
Con pubblicazione nella G.U. dell’Unione Europea (6 giugno 2014) la Formaldeide dal 1 aprile 2015 viene considerata: cancerogena 1/B (Regolamento U.E. n.605/2014 del 5 giugno 2014).
In pratica, ove la formaldeide sia presente e ci siano lavoratori esposti, bisognerà procedere a rivedere il Documento di Valutazione di Rischio secondo le indicazioni del Titolo IX del D.Lvo 81/08 (protezione da agenti cancerogeni e mutageni) e dare corso a tutti gli obblighi previsti (sistemi chiusi, eliminazione o riduzione del rischio, registro esposti, sorveglianza sanitaria …), tutto entro il primo aprile.
Perché anche gli zuccheri, aminoacidi e proteine sono aggiunti in alcuni vaccini?
Queste sostanze possono essere aggiunte come stabilizzanti. Essi consentono di proteggere il vaccino da condizioni avverse come il processo di liofilizzazione.
Gli Stabilizzatori aggiunti ai vaccini comprendono: zuccheri come saccarosio e lattosio, amminoacidi come glicina o il sale monosodico dell’acido glutammico e proteine come l’albumina sierica umana o la gelatina. Zuccheri, amminoacidi e proteine non sono unici per vaccini e si incontrano nella vita quotidiana nella dieta e sono componenti che sono corpo naturalmente.
***SAREBBE INTERESSANTE INDAGARE SULLE EVENTUALI PREDISPOSIZIONI E INTOLLERANZE A SOSTANZE COME IL LATTOSIO***
Perché alcuni vaccini contengono conservanti ?
I Conservanti vengono aggiunti in alcune formulazioni per prevenire la crescita di batteri o funghi che potrebbero contaminare il vaccino durante il suo utilizzo.
***ESEMPIO DI CONTAMINAZIONI?***
Il CDC ha pubblicato sul proprio sito web ( deposto questo post e poi STRANAMENTE ELIMINATO). Clicca qui per una copia archiviata internet ), un dato sconcertante:
“ben 30 milioni di americani potrebbero essere a rischio di sviluppare il cancro a causa di vaccino antipolio contaminato con Simian Virus 40 (SV40) in alcune specie di scimmie”.
Nella produzione di vaccini virali, il virus può essere coltivato in cellule. Queste cellule hanno bisogno di una fonte di alimentazione, che in alcuni casi può essere fornita dal siero bovino fetale.
L’organo FDA è blando nel riportare queste affermazioni in realtà contrastanti con l’altra parte della comunità scientifica.
Di seguito gli allegati di riferimento.
Secondo l’Environmental Defense (precedentemente noto come l’Environmental Defense Fund), tutti gli ingredienti dei vaccini sono tossici, cancerogeni o potenzialmente dannosi per la pelle. Questi agiscono dando problematiche gastrointestinali, polmonari, al sistema immunitario e neurologico.
MSG, antigelo,fenolo (usato come disinfettante), formaldeide (che causano il cancro e usato per imbalsamare), alluminio (associata con la malattia e le convulsioni di alzheimer), glicerina (tossico per i reni, il fegato, può causare danni ai polmoni, danni gastrointestinali e morte ), piombo, cadmio, solfati, le proteine del lievito, antibiotici, acetone (utilizzato in acetone), neomicina (antibiotico) e streptomicina(antibiotici). Thimerosal (più tossico del mercurio, un conservante ancora in uso in molti vaccini,può causare gravi danni neurologici così come altre in pericolo di vita malattie autoimmuni). Sappiamo la coltura di questi vaccini? La conosciamo?
Questi vaccini sono coltivati su animali (organi) o tessuti umani, come i reni di scimmia, embrioni di pollo, siero di vitello, cellule diploidi umane (gli organi sezionati di feti abortiti), sangue di maiale, sangue di cavallo e il cervello di coniglio.
Il potenziale di contaminazione da non rilevati, gravissime virus animali è molto elevato.
Con determinati elementi incredibilmente tossici elencati di seguito a cui un bambino è sottoposto diverse volte,non c’è da meravigliarsi sui risultati di questi sondaggi.
I medici sono perplessi certamente. Tutti conosciamo gli elementi attivi contenuti nei vaccini.
I bugiardini parlano chiaro.
Di seguito allegati e fonti di riferimento su bugiardini e due intere sezioni dedicate al Mercurio e l’Alluminio
Il problema di queste affermazioni è che non sono supportate da prove. Quando guardiamo i dati effettivi, vediamo altro. Benché molte persone siano morte a causa della pertosse nella prima parte del 1900, dal momento dell’introduzione del vaccino, il tasso di morte negli Stati Uniti è diminuito di oltre il 90 per cento. Utilizzando la fonte che è stata il riferimento per affermare tutto questo sul giornale Pediatrics, vediamo che il calo delle morti dalla vetta è stato di circa il 92 per cento prima dell’introduzione del vaccino DTP…
Currently, the increasing numbers of vaccine administrations are associated with increased reports of adverse vaccine reactions. Whilst the general adverse reactions including allergic reactions caused by the vaccine itself or the vaccine components, are rare, they can in some circumstances be serious and even fatal. In accordance with many IgE-mediated reactions and immediate-type allergic reactions, the primary allergens are proteins. The proteins most often implicated in vaccine allergies are egg and gelatin, with perhaps rare reactions to yeast or latex. Numerous studies have demonstrated that the injectable influenza vaccine can be safely administered, although with appropriate precautions, to patients with severe egg allergy, as the current influenza vaccines contain small trace amounts of egg protein. If an allergy is suspected, an accurate examination followed by algorithms is vital for correct diagnosis, treatment and decision regarding re-vaccination in patients with immediate-type reactions to vaccines. Facilities and health care professionals should be available to treat immediate hypersensitivity reactions (anaphylaxis) in all settings where vaccines are administered.
Meccanismi di tossicità adiuvante alluminio e autoimmunità nella popolazione pediatrica.
http://www.ncbi.nlm.nih.gov/pubmed/22235057
Coadiuvanti in alluminio: sono sicuri?
Astratto
L’alluminio è la neurotossina più utilizzata come adiuvante nei vaccini. Nonostante quasi 90 anni di uso diffuso,la scienza è ancora allo scuro sul meccanismo d’azione della medesima. Ricerche sperimentali, tuttavia, mostrano chiaramente che i coadiuvanti d’alluminio possono indurre a gravi malattie immunologiche. L’alluminio è un rischio per l’autoimmunità e le complicanze neurologiche associate e può quindi avere conseguenze per la salute negative (profonde e diffuse).
http://www.ncbi.nlm.nih.gov/pubmed/21568886
Ci sono effetti negativi sul sistema nervoso centrale di coadiuvanti alluminio utilizzati nei vaccini e immunoterapia?
http://www.ncbi.nlm.nih.gov/pubmed/25428645
BIOPERSISTENZA E TRASLOCAZIONE CEREBRALE DELL’ALLUMINIO UTILIZZATO COME ADIUVANTE NEI VACCINI
L’utilizzo di antigeni altamente puri per migliorare la sicurezza del vaccino ha portato a ridurre l’immunogenicità del vaccino e l’efficacia. Questo ha portato alla necessità di utilizzare adiuvanti per migliorare l’immunogenicità del vaccino. L’adiuvante ideale dovrebbe massimizzare l’immunogenicità del vaccino senza compromettere la tollerabilità e la sicurezza.
Il primo vaccino del mio bambino; cosa consiglia il Center for Disease control?
Per quali malattie vacciniamo?
Quali sono i vaccini obbligatori?
Cos’è il consenso informato?
Come siamo sicuri che il nostro bimbo non abbia possibili reazioni dopo?
E’ giusto avere risposta a tutte queste lecite domande.Oramai con gli studi tradotti abbiamo capito una cosa importantissima;le malattie sono state debellate non dai vaccini ma da tutt’altre condizioni presenti in quel momento e a dirlo sono grafici e studi reali .
Su queste malattie ne sono state raccontate di ogni e ancora non si è capita -volutamente- la differenza tra VACCINAZIONE e IMMUNIZZAZIONE.
Questi due termini sono diversi e raccontano due stati e condizioni diverse,è bene saperlo.Vaccinare non significa immunizzare e di seguito leggeremo direttamente la traduzione delle affermazioni e consigli dell’Ente CDC in merito alla prima vaccinazione,cosa bisogna sapere PRIMA.
Ricordiamo intanto facendo qualche passo indietro alcune informazioni di base…
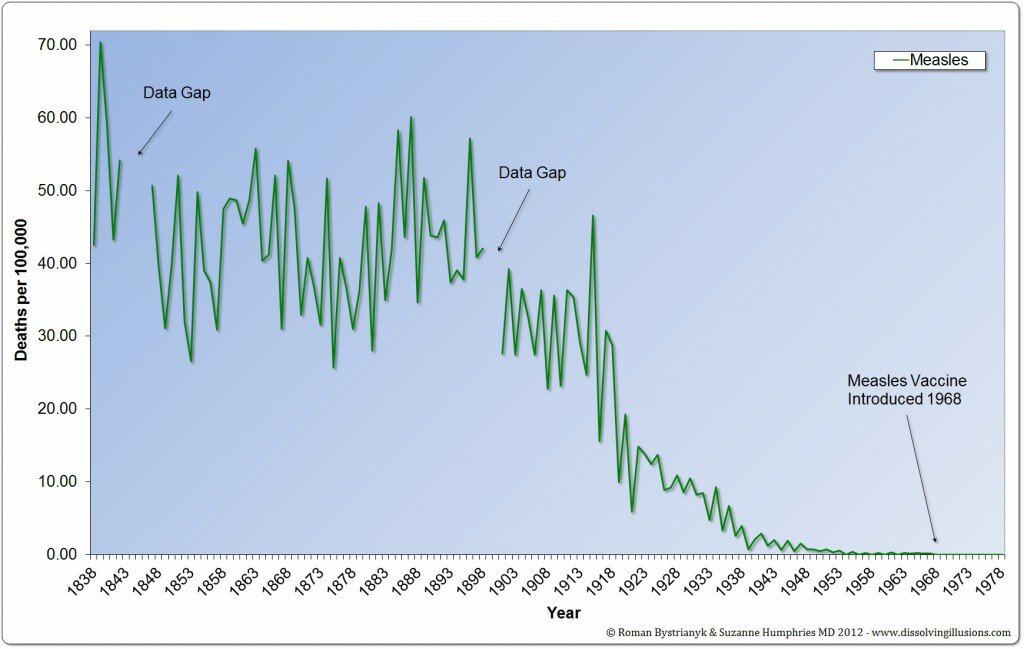
A proposito di tabelle e grafici
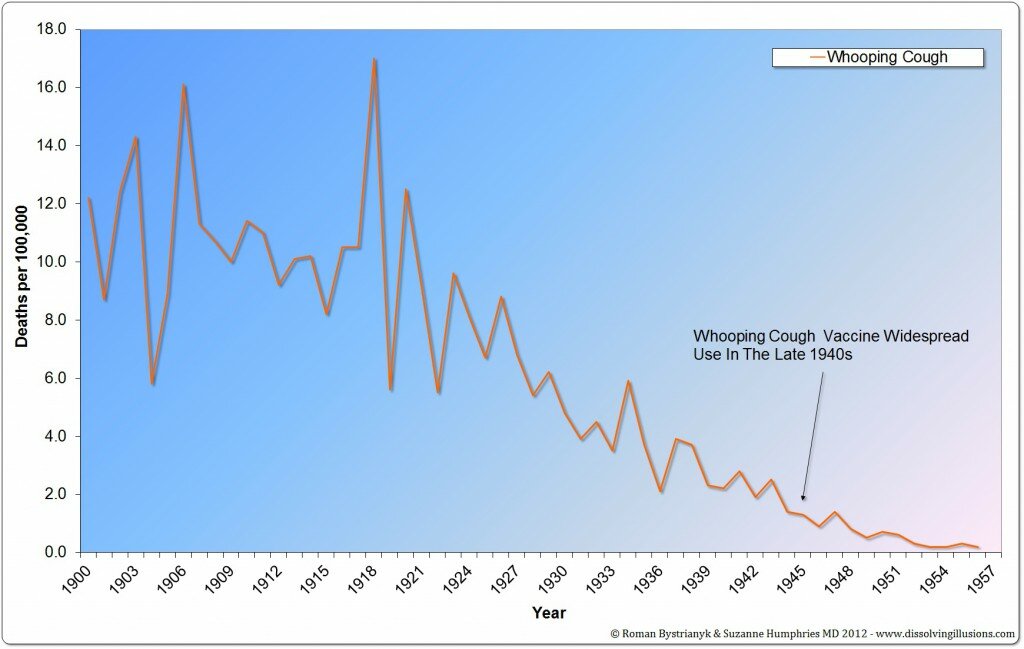
IL 92% DEL TASSO DI MORTE SI E’ RIDOTTO PRIMA DELL’INTRODUZIONE DEL VACCINO DTP
Il numero effettivo di morti dal momento dell’introduzione del vaccino DTP era di circa 1.200 e non i 5.000 e 10.000 varie volte citati.
Un punto ulteriormente importante da notare è la riduzione della mortalità (per pertosse) ogni anno, a prescindere dall’introduzione di questo vaccino.Guardando i grafici evince questa realtà,l’introduzione del vaccino,non ha portato a nessuna tendenza al ribasso,questo fenomeno era fortunatamente iniziato prima.
Un’altra serie di dati a partire dall’inizio del 20° secolo, mostrano la mancata riduzione di mortalità dei vaccini ancora più drammaticamente. Qui si può vedere che il tasso di mortalità era sceso di oltre il 98%prima dell’uso nazionale della vaccinazione DTP nel 1950.
IL TASSO DI MORTALITA’ ERA SCESO OLTRE IL 98% PRIMA DELL’USO DEL VACCINO DTP
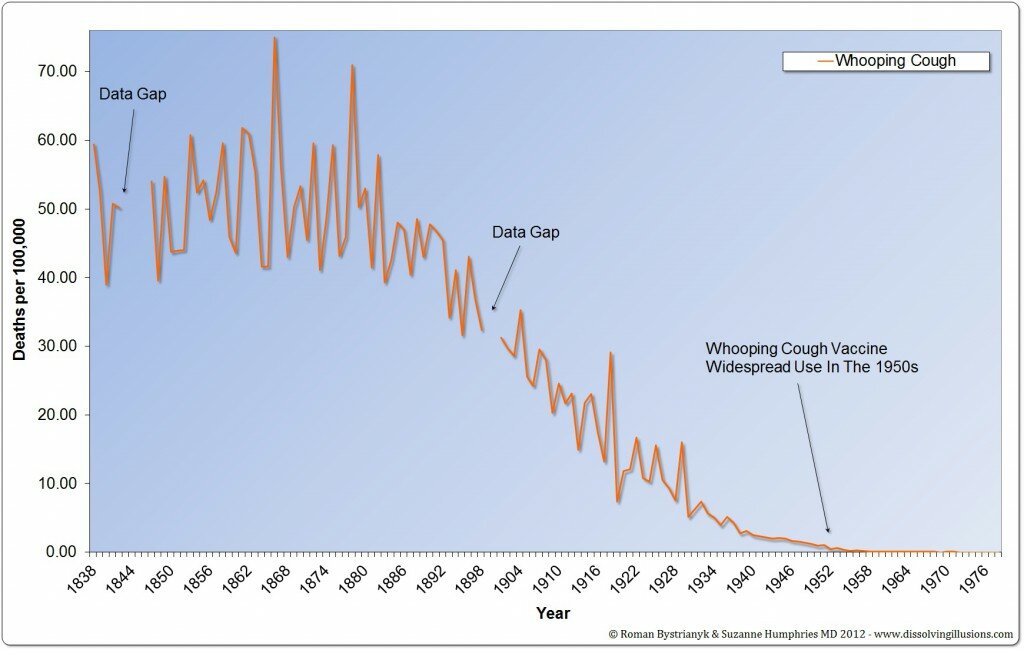
l’Inghilterra iniziò con il mantenere le statistiche nel 1838 (62 anni prima erano state riunite le statistiche ufficiali statunitensi). Guardando questi dati, possiamo notare, che il tasso di mortalità per le malattie infettive era alto nel corso del 1800 ed è diminuito dalla metà del 1800 alla metà del 1900 arrivando quasi a zero. Guardando i dati di mortalità per pertosse in Inghilterra, i decessi sono diminuiti di oltre il 99% prima di qualsiasi vaccino.
I DECESSI DIMINUIRONO DI OLTRE IL 99% PRIMA DELL’INTRODUZIONE IN INGHILTERRA DI QUALSIASI VACCINO
Nel caso del morbillo, vediamo una riduzione del tasso di mortalità di quasi il 100 %
I tassi di mortalità sono diminuiti prima della vaccinazione. Nel caso della scarlattina e di altre malattie infettive, i decessi sono diminuiti quasi a zero, senza alcuna vaccinazione su larga scala.
Purtroppo, queste credenze errate, hanno da sempre portato a fidarsi della vaccinazione, credendo a tutto ciò che ci viene da sempre propinato,non chiedendo e non visionando mai dati,grafici o tabelle come queste.
Da sempre si crede nella virtù della vaccinazione,la quale ha debellato le malattie,senza riflettere su altri fattori,che in realtà hanno causato la mortalità a diminuire.
Questi fattori sono l’igiene, i servizi igienico-sanitari, la nutrizione, il diritto del lavoro,l’ elettricità, la clorazione, la refrigerazione, la pastorizzazione, e molti altri aspetti che oggi generalmente diamo per scontati come parte della vita moderna.
Il miglioramento del tasso della mortalità aveva molto poco a che fare con la medicina.
Un rapporto 1977 ha stimato che, nel migliore dei casi, circa il 3% del declino della mortalità per malattie infettive potrebbe essere attribuito alle cure mediche moderne.
Cosa non viene detto inoltre sulla diffusione delle malattie e i soggetti vaccinati?
I funzionari della sanità pubblica conoscono il vero: i soggetti vaccinati di recente sono “causa” di diffusione delle malattie.
ULTIMAMENTE I MEDIA E I GIORNALI HANNO INIZIATO UNA VERA E PROPRIA CACCIA ALLE STREGHE AFFERMANDO CHE IL CALO DELLE VACCINAZIONI E I NON VACCINATI, SIANO RESPONSABILI DELLA DIFFUSIONE DI PATOLOGIE PER LE QUALI TANTO SI E’ LOTTATO PER L’ERADICAZIONE.
Di seguito il link della notizia tradotta e riportata dal GLOBE NEWSWIRE (Washington).
Per rispondere a tutte le domande iniziali,poniamo di seguito i link di accesso ad una lettura che serivirà a chiarire da ogni angolazione questo vasto argomento.
Malattie e relativi vaccini
Approfondimenti e Aggiornamenti con fonti e allegati La malattia “POLIOMELITE” La poliomielite è una malattia infettiva causata da tre diversi virus (poliovirus 1, 2 e 3) che sono capaci di rimanere attivi nell’ambiente anche per anni dopo la loro espulsione dall’individuo infetto…
QUALI SONO I TEST DA EFFETTUARE PRIMA DI VACCINARE? ESISTONO? IN COSA CONSISTONO? POSSONO FARSI ANCHE DOPO IL VACCINO?
Il problema dei test di laboratorio prima e dopo unavaccinazione pediatrica è un problema serio e spesso ricevo richieste di questo tipo da parte dei genitori che sono incerti se vaccinare o meno i loro figli e, per avere elementi più precisi per decidere, vorrebbero sottoporre i loro figli a dei test ematochimici allo scopo di conoscere, come sarebbe logico e giusto, le loro condizioni metaboliche e immunitarie.
Cosa realmente sappiamo del Consenso Informato? .Veniamo realmente messi a conoscenza di tutte le informazioni disponibili sulla nostra salute ? Il Medico deve informare sempre il paziente ad esempio sull’effetto di un farmaco,sui suoi effetti collaterali, su una terapia,tutto insomma; Non farlo costituisce REATO..
QUALI SONO I VACCINI OBBLIGATORI E I FACOLTATIVI? I vaccini obbligatori in Italia sono solo Difterite, Tetano, Polio ed Epatite B, ad esclusione del Veneto che ha eliminato l’obbligo dal 2008. Il vaccino esavalente contiene anche: anti-Haemophilus tipo b (Hib) coniugato e antipertossico (componenti acellulari). Solo in Veneto è stato eliminato l’obbligo dal 2008..
Tornando a ciò che dichiara il CDC “queste malattie sono molto meno comuni di quelli di una volta”.
Ne siamo proprio sicuri? Ovviamente un Organo così importante non potrebbe sconsigliare la vaccinazione; egli deve continuare a seguire il protocollo standard,consigliando la vaccinazione,ovvero;
I VACCINI CONSIGLIATI SONO:
Vaccino
Numero di dosi
Età consigliata
Altre informazioni
DTaP (difterite, tetano, pertosse)
5
2 mesi, 4 mesi, 6 mesi, 15-18 mesi, 4-6 anni
Alcuni bambini non dovrebbero ricevere il vaccino della pertosse. Questi bambini possono ottenere un vaccino chiamato DT (difterite e tetano).
Epatite B
3
Nascita, 1-2 mesi, 6-18 mesi
Polio
4
2 mesi, 4 mesi, 6-18 mesi, 4-6 anni
Hib (Haemophilus influenzae tipo b)
3 o 4
2 mesi, 4 mesi, (6 mesi), 12-15 mesi
PCV13 (pneumococco)
4
2 mesi, 4 mesi, 6 mesi, 12-15 mesi
L’operatore sanitario potrebbe offrire alcuni di questi vaccini come “vaccini combinati” (diversi vaccini somministrati nella stessa dose).
Alcuni bambini non dovrebbero avere alcun vaccino!
Un bambino che non è in ottimale stato di salute,non può essere vaccinato.
Un bambino che ha avuto una reazione allergica dopo la somministrazione di un vaccino non dovrebbe ottenere alcuna vaccinazione in seguito.
Un bambino che ha una grave allergia ad una sostanza contenuta nei vaccini, ovviamente non dovrebbe essere vaccinato. Alcuni di questi vaccini contengono neomicina, streptomicina, lievito, lattosio, saccarosio, o lattice.
***OSSERVAZIONI***SE QUESTE AFFERMAZIONI ESISTONO, SONO DATI DI FATTO.
PARTIAMO DAL PRESUPPOSTO CHE I DANNEGGIATI ESISTONO E CHE NON SONO UNO SU UN MILIONE.I DATI SONO DIVERSI E MOLTISSIMI PROFESSIONISTI STANNO PRENDENDO COSCIENZA SU QUESTO ARGOMENTO.
Come conosciamo la situazione completa dello stato di salute dei nostri figli (patologie/allergie esistenti etc) senza effettuare una corretta anamnesi?
COME FACCIAMO A CONOSCERE LE EVENTUALI ALLERGIE -SE PRESENTI- SENZA EFFETTUARE DEI TEST DI LABORATORIO PRIMA? COME MAI IL PEDIATRA NON METTE A CONOSCENZA NOI GENITORI DI QUESTE IMPORTANTI QUESTIONI?
Il CDC CONSIGLIA la massima attenzione:
Vaccino DTaP , se il vostro bambino ha avuto una delle seguenti reazioni dopo una precedente dose di vaccino.
Una danno al cervello o una malattia del sistema nervoso entro 7 giorni.
Pianto inconsolabile per 3 ore o più.
Un collasso,o crisi epilettiche,spasmi.
Febbre di oltre i 39.
Vaccino antipolio ,se il vostro bambino ha una grave allergia alla neomicina, streptomicina o polimixina B.
Vaccino contro l’epatite B , se il vostro bambino ha una grave allergia al lievito.
Vaccino PCV13 , se il vostro bambino ha una grave allergia al lievito.
I rischi di una reazione al vaccino, non sono bassi. Ricordiamo che i Vaccini sono Farmaci e come tali,possono causare dei danni e degli effetti indesiderati.
Altri vaccini per l’infanzia sono stati associati a questi problemi aggiuntivi:
DTaP Vaccino
Febbre alta.
Stanchezza o scarso appetito (fino a 1 bambino su 10); vomito (fino a 1 bambino in 50); gonfiore del tutto il braccio o la gamba per 1-7 giorni (fino a 1 bambino in 30) – di solito dopo il 4 ° o 5 ° dose.
Non smettere di piangere per 3 ore o più (fino a 1 bambino su 1.000); febbre oltre i 39° (1 bambino su 16.000).
Gravi problemi: convulsioni lungo termine, coma,e danni permanenti al cervello sono stati riportati a seguito della vaccinazione DTaP.
Vaccino pneumococcico
Perdita di Sonno o perdita di appetito (circa 1 bambino su 2 o 3);
Febbre alta e problemi vari(circa 1 bambino su 20).
Reazioni che potrebbero accadere dopo qualsiasi vaccino:
Svenimenti brevi possono accadere dopo ogni procedura medica, tra cui una vaccinazione. Seduti o sdraiati per circa 15 minuti può aiutare a prevenire svenimenti, e le lesioni causate da una caduta.
Grave dolore alla spalla e una ridotta gamma di movimento nel braccio in cui si vaccina,può accadere,dopo una vaccinazione,ma dovrebbe essere raro.
Gravi reazioni allergiche di un vaccino: se si dovessero verificarsi, sarebbe solitamente entro pochi minuti ad alcune ore dopo la vaccinazione.
“Come con qualsiasi medicinale, c’è una possibilità molto remota di un vaccino che causa un grave infortunio o morte”.
Per ulteriori informazioni, visitare il sito web di CDC Vaccine Safety .
In caso di Reazione,Cosa si dovrebbe fare?
Se accade una qualsiasi reazione, successiva alla vaccinazione,si chiama immediatamente il pronto soccorso. In caso contrario, chiamare il medico.
In seguito, la reazione deve essere segnalato al Vaccine Adverse Event Reporting System (VAERS). Il medico potrebbe presentare questa relazione, o si può fare da soli attraverso il sito web VAERS , o chiamando 1-800-822-7967.
Il VAERS non dà consigli medici.
Ricordiamo lo stesso procedimento in Italia attraverso segnalazione al sistema di FARMACOVIGILANZA.
The National Vaccine Injury Compensation Program
Il National Vaccine Injury Compensation Program (VICP) è un programma federale creato per compensare le persone danneggiate da vaccini.
ASTRATTO DAL SITO CDC
” Any vaccine can cause side effects”
Cosa vuol dire?
“Ogni vaccino può causare effetti indesiderati”; di seguito il testo e la fonte.
Any vaccine can cause side effects. For the most part these are minor (for example, a sore arm or low-grade fever) and go away within a few days. Listed below are vaccines licensed in the United States and side effects that have been associated with each of them. This information is copied directly from CDC’s Vaccine Information Statements, which in turn are derived from the Advisory Committee on Immunization Practices (ACIP) recommendations for each vaccine….
CONTINUIAMO A RACCONTARCI LA FAVOLA DELL’OMISSIONE DELLA REALTA’.
TUTTO VIENE SEMPRE NEGATO,OMESSO,TACIUTO.
SE ESISTE UN SISTEMA DI SEGNALAZIONE DI REAZIONI AVVERSE,SE QUESTO è STATO CREATO E CONCEPITO,VUOL DIRE CHE PURTROPPO LE REAZIONI AVVERSE CI SONO ,CI SONO STATE E CI SARANNO E CHE TALI NON SONO UNA SU UN MILIONE.
I DATI DICONO DIVERSAMENTE,BASTI GUARDARE L’AUMENTO DEI TASSI DELL’AUTISMO.
A TUTTO C’E’ UNA RISPOSTA,BASTA SAPERE DOVE GUARDARE.
Morbillo? Nessun decesso negli ultimi dieci anni. Sono più di 100 le segnalazioni di reazioni avverse anche letali, avvenute dopo la somministrazione del vaccino. 6 Marzo 2015 Ultimamente troviamo bufera in tutto il web. La cattiva informazione e la manipolazione dei dati, fanno da padrone..
L’American Academy of Pediatrics (AAP) non raccomanda la vaccinazione di routine dei bambini tra i 2 mesi e i 10 anni di età a meno che non ci siano dei rischi di malattia meningococcica. Aggiornamento delle Linee Guida Internazionali Linee guida sul vaccino anti-meningococco e alcune riflessioni sulla meningite da meningococco C Sono state aggiornate a cura […]
FARMACI RITIRATI DAL COMMERCIO e/o OGGETTO DI MONITORAGGIO
QUARTA PARTE
IL FARMACO KILLER CHE CURA L’ARTRITE MA DISTRUGGE IL FEGATO: “VIETATO NEGLI USA, NON IN ITALIA”
lunedì 23 giugno 2014
ROMA – Negli Stati Uniti è già stato vietato da tempo, ma nel nostro paese un farmaco che dovrebbe curare l’artrite, ma può avere effetti devastanti sul fegato, è ancora in vendita.
Si tratta di Arava, un prodotto per la cura dell’artrite reumatoide che, a causa di un principio attivo, il leflunomide, può avere effetti collaterali devastanti per il fegato.
Per questo motivo gli Stati Uniti lo hanno già vietato, ma in Italia la vendita è autorizzata dal 2009.
I medici considerano Arava ben più pericoloso di Aulin, il celebre antidolorifico vietato dopo aver scoperto che, se assunto in grandi quantità, aveva effetti molto simili.
C’è anche un caso di morte dovuta a questo farmaco: quella di Manuela, 18enne venezianamorta nel2002pernecrosi del fegato. La ragazza, che aveva dolori ossei soprattutto alle piccole articolazioni, si sottopose al trattamento con Arava per 10 mesi. Durante la cura non migliorò, ma continuò il trattamento, fino alla morte, avvenuta in ospedale.
Il referto medico ha accertato che Manuela è morta per necrosi al fegato, e sono stati gli stessi sanitari del’Asl 13 a relazionare al ministero della Sanità l’uso del farmaco collegandolo al decesso della giovane.
La famiglia non si è data pace, ha passato diverso tempo a cercare un medico che potesse opporsi ad un’Asl su un farmaco ancora legalizzato.
La sentenza del giudice di Dolo ha dato però ragione ai famigliari di Manuela condannando l’azienda, e indirettamente il medico, a risarcire la famiglia con 500 mila euro.
Tuttora nel prontuario farmaceutico. Tribunale: “va ritirato dal mercato”.
Nel 2002 ha provocato necrosi del fegato e morte di una diciottenne.
L’Italia è stato l’ultimo paese europeo a vietare la vendita del prodotto
ROMA- Effetti collaterali terribili, che in un caso particolare di una diciottenne di Dolo (provincia di Venezia), nel 2002 hanno portato al decesso.
Stiamo parlando di Arava, un farmaco che, secondo quanto riporta Vip, è stato vietato negli Stati Uniti proprio a causa di questi effetti collaterali che colpirebbero la necrosi del fegato.
La vicenda della ragazza è tornata d’attualità dopo che nei giorni scorsi il Tribunale di Dolo ha emesso il verdetto, dichiarando la pericolosità del medicinale e stabilendo la somma del risarcimento che la società farmaceutica dovrà versare ai genitori della ragazzina morta nel 2002.
Il farmaco in teoria è utilizzato per la cura dell’artrite reumatoide e psoriasica.
Aveva avuto il via libera della Cuf, la Commissione unica del farmaco presieduta dal ministro della Sanità, ed è tuttora nel prontuario farmaceutico in Italia. Nonostante la sua pericolosità, saremo uno degli ultimi paesi dell’Unione Europea a ritirarlo dal mercato.
La conferma dell’autorizzazione alla vendita, nonostante negli Usa fosse rimasto vietato, era stata ottenuta nel 2009.
Il problema principale di Arava è che presenta al suo interno un principio attivo, il leflunomide, un immunosoppressore necessario a tenere basso il numero di globuli bianchi nei malati di artrite reumatoide attiva, e la psoriasica attiva, due malattie che portano la prima infiammazioni delle articolazioni e la seconda, in aggiunta, la comparsa di chiazze rosse di desquamazione sulla pelle.
Tornando alla vicenda della ragazza, la sentenza del giudice di Dolo ha dato ragione ai familiari, condannando l’azienda e indirettamente il medico (che le aveva prescritto il farmaco), a risarcire la famiglia con 500 mila euro.
Arava 20 mg compresse rivestite con film Riassunto delle Caratteristiche del Prodotto
INDICE DELLA SCHEDA
01.0 DENOMINAZIONE DEL MEDICINALE
02.0 COMPOSIZIONE QUALITATIVA E QUANTITATIVA
03.0 FORMA FARMACEUTICA
04.0 INFORMAZIONI CLINICHE
05.0 PROPRIETÀ FARMACOLOGICHE
06.0 INFORMAZIONI FARMACEUTICHE
07.0 TITOLARE DELL’AUTORIZZAZIONE ALL’IMMISSIONE IN COMMERCIO
08.0 NUMERI DELLE AUTORIZZAZIONI ALL’IMMISSIONE IN COMMERCIO
09.0 DATA DELLA PRIMA AUTORIZZAZIONE/RINNOVO DELL’AUTORIZZAZIONE
10.0 DATA DI REVISIONE DEL TESTO
01.0 DENOMINAZIONE DEL MEDICINALE ARAVA 20 MG COMPRESSE RIVESTITE CON FILM
02.0 COMPOSIZIONE QUALITATIVA E QUANTITATIVA
Ogni compressa contiene 20 mg di leflunomide. Eccipienti con effetti noti: Ogni compressa contiene 72 mg di lattosio monoidrato. Per l’elenco completo degli eccipienti, vedere paragrafo 6.1.
03.0 FORMA FARMACEUTICA
Compressa rivestite con film. Compressa rivestita con film triangolare, da giallastra a ocra, con impresso ZBO su un lato.
04.0 INFORMAZIONI CLINICHE
04.1 Indicazioni terapeutiche
La leflunomide è indicata nel trattamento di pazienti adulti affetti da: • artrite reumatoide attiva, come farmaco antireumatico in grado di modificare il decorso della malattia (DMARD – Disease–Modifying Antirheumatic Drug), • artrite psoriasica attiva. Un recente o concomitante trattamento con DMARD epatotossici o ematotossici (ad esempio metotrexato) può portare ad un aumentato rischio di reazioni avverse gravi; quindi, prima di iniziare una terapia con leflunomide si deve fare un’attenta valutazione in termini di rischio/beneficio. Inoltre, il passaggio da leflunomide ad altri DMARD senza seguire la procedura di washout (vedere paragrafo 4.4) può anche aumentare il rischio di reazioni avverse gravi anche per un lungo periodo dopo tale passaggio.
04.2 Posologia e modo di somministrazione
Il trattamento deve essere iniziato e controllato da specialisti esperti nel trattamento dell’artrite reumatoide e dell’artrite psoriasica. Alanina aminotransferasi (ALT) o glutammico piruvico transaminasi sierica (SGPT) e un test ematologico completo, inclusa una formula leucocitaria differenziata e una conta piastrinica, devono essere controllati simultaneamente e con la stessa frequenza: • prima dell’inizio della terapia con leflunomide, • ogni 2 settimane durante i primi 6 mesi di terapia, e • successivamente ogni 8 settimane (vedere paragrafo 4.4).
Posologia • Artrite reumatoide: la terapia con leflunomide viene di solito iniziata con dose di carico di 100 mg una volta al giorno, per 3 giorni. Evitare di somministrare la dose di carico può diminuire il rischio di reazioni avverse (vedere paragrafo 5.1). La dose di mantenimento raccomandata va da 10 a 20 mg una volta al giorno in funzione della gravità (attività) della malattia. • Artrite psoriasica: la terapia con leflunomide inizia con una dose di carico di 100 mg una volta al giorni per 3 giorni. La dose di mantenimento raccomandata è di 20 mg di leflunomide una volta al giorno (vedere paragrafo 5.1). Normalmente l’effetto terapeutico si manifesta dopo 4–6 settimane di trattamento e può ulteriormente incrementare entro 4–6 mesi. Non è previsto alcun aggiustamento del dosaggio in pazienti affetti da insufficienza renale lieve. Non è necessario un aggiustamento del dosaggio nei pazienti di età superiore ai 65 anni. Popolazione pediatrica Arava non è raccomandato nei pazienti di età inferiore ai 18 anni poichè l’efficacia e la sicurezza nell’artrite reumatoide giovanile (ARJ) non sono state stabilite (vedere paragrafi 5.1 e 5.2).
Modo di somministrazione Le compresse di Arava devono essere assunte intere con sufficiente quantità di liquido. Il grado di assorbimento della leflunomide non è influenzato dall’assunzione di cibo.
04.3 Controindicazioni
• Ipersensibilità al principio attivo (specialmente precedenti di sindrome di Stevens–Johnson, necrolisi epidermica tossica, eritema multiforme) o ad uno qualsiasi degli eccipienti elencati al paragrafo 6.1.
• Pazienti con insufficienza epatica.
• Pazienti affetti da immunodeficienza grave (ad esempio AIDS).
• Pazienti con funzionalità midollare significativamente compromessa o con anemia, leucopenia, neutropenia o trombocitopenia gravi, ad eziologia diversa dall’artrite reumatoide o dall’artrite psoriasica.
• Pazienti con infezioni gravi, (vedere paragrafo 4.4).
• Pazienti con insufficienza renale da moderata a grave, perché in tale gruppo di pazienti non sono disponibili sufficienti esperienze cliniche.
• Pazienti con ipoproteinemia grave, ad esempio nella sindrome nefrosica.
•Donne in gravidanza o donne in età feconda che non facciano uso di metodi contraccettivi affidabili durante il trattamento con leflunomide.
Dopo sospensione del trattamento con leflunomide, la gravidanza è controindicata sino a che le concentrazioni plasmatiche del metabolita attivo risultino superiori a 0,02 mg/l (vedere paragrafo 4.6). Prima di iniziare il trattamento con leflunomide, si raccomanda di escludere una gravidanza. • Donne che allattano (vedere paragrafo 4.6).
04.4 Speciali avvertenze e precauzioni per l’uso
La concomitante somministrazione di DMARD, epatotossici o ematotossici (ad esempio metotrexato) non è consigliabile. Il metabolita attivo della leflunomide, A771726, ha una lunga emivita, solitamente tra 1 e 4 settimane.
Si potrebbero avere effetti indesiderati gravi (ad esempio epatotossicità, ematotossicità o reazioni allergiche, vedere sotto), anche se il trattamento con leflunomide è stato interrotto. Quindi, quando dovessero manifestarsi tali reazioni tossiche o se per qualsiasi altra ragione dovesse essere necessario eliminare A771726 rapidamente dal corpo, deve essere seguita la procedura di washout.
Tale procedura può essere ripetuta se clinicamente necessario. Per le procedure di washout e per le altre azioni raccomandate in caso di una gravidanza programmata o inaspettata vedere paragrafo 4.6.
Reazioni epatiche
Rari casi di grave danno epatico, inclusi i casi letali, sono stati riportati in corso di trattamento con leflunomide. Molti di questi casi si sono verificati entro i primi 6 mesi di trattamento. Trattamenti concomitanti con altri farmaci epatotossici erano frequentemente presenti. Si ritiene essenziale che le raccomandazioni di controllo siano attentamente seguite.
I livelli di ALT (SGPT) devono essere controllati prima di iniziare il trattamento con leflunomide e con la stessa frequenza del test ematologico completo (ogni 2 settimane) durante i primi 6 mesi di terapia e successivamente ogni 8 settimane.
Per aumenti dei livelli di ALT (SGPT) da 2 a 3 volte il limite superiore al normale, la riduzione della dose di Arava da 20 a 10 mg deve essere presa in considerazione e deve essere effettuato un monitoraggio settimanale. Se l’aumento dei livelli di ALT (SGPT) maggiore di 2 volte il limite superiore al normale persiste o se l’aumento è maggiore di 3 volte, la leflunomide deve essere sospesa e deve essere iniziata la procedura di washout.
Si raccomanda che il monitoraggio degli enzimi epatici sia effettuato dopo l’interruzione del trattamento con leflunomide, fino a che i livelli degli enzimi epatici siano normalizzati. Data la possibilità di accentuazione degli effetti epatotossici, si raccomanda di astenersi dall’assunzione di bevande alcoliche nel corso del trattamento con leflunomide. Poiché il metabolita attivo della leflunomide, A771726, presenta un elevato legame con le proteine plasmatiche e viene eliminato attraverso il metabolismo epatico e la secrezione biliare, i livelli plasmatici di A771726 possono aumentare nei pazienti con ipoproteinemia. Arava è controindicato in pazienti con ipoproteinemia o insufficienza epatica gravi (vedere paragrafo 4.3).
Reazioni ematologiche
Unitamente ai livelli di ALT, un test ematologico completo inclusa la formula leucocitaria e le piastrine, deve essere eseguito prima dell’inizio del trattamento, nonché ogni 2 settimane per i primi 6 mesi di terapia e successivamente ogni 8 settimane.
Nei pazienti con anemia preesistente, leucopenia, e/o trombocitopenia come pure nei pazienti con ridotta funzionalità del midollo osseo o che sono a rischio di soppressione dell’attività del midollo osseo il rischio di alterazioni ematologiche è aumentato.
Se dovessero manifestarsi tali effetti, si deve prendere in considerazione un washout (vedere sotto) per ridurre i livelli plasmatici di A771726. In caso di reazioni ematiche gravi, inclusa la pancitopenia, devono essere sospesi Arava e qualunque altro trattamento mielosoppressivo concomitante e si deve iniziare una procedura di washout di Arava.
Associazione con altri trattamenti
L’uso di leflunomide con gli antimalarici utilizzati nelle malattie reumatiche (per esempio clorochina e idrossiclorochina), l’oro somministrato per via intramuscolare o orale, la D–penicillamina, l’azatioprina ed altri immunosoppressori compresi gli inibitori del TNF–alfa non è stato ancora adeguatamente studiato in studi randomizzati (ad eccezione del metotrexato – vedere paragrafo 4.5).
Non si conosce il rischio associato ad una terapia in associazione, in particolare per un trattamento a lungo termine. Poiché tale terapia può causare tossicità additiva o anche sinergica (ad esempio epato– o ematotossicità), l’associazione con un altro DMARD (ad esempio metotrexato) non è consigliabile. Si deve usare precauzione quando leflunomide è somministrata con altri farmaci come i FANS metabolizzati da CYP2C9, come la fenitoina, la warfarina, il fenprocumone e la tolbutamide.
Passaggio ad altre terapie
Poiché la leflunomide rimane a lungo nel corpo, il passaggio ad un altro DMARD (ad esempio metotrexato) senza praticare la procedura di washout (vedere sotto) può aumentare la possibilità di rischi addittivi anche per un lungo periodo di tempo dopo la sostituzione (cioè interazioni cinetiche, tossicità d’organo).
Analogamente, un recente trattamento con farmaci epatotossici o ematotossici (ad esempio metotrexato) può portare ad un aumento degli effetti indesiderati; quindi, l’inizio di un trattamento con leflunomide deve essere attentamente valutato per quanto riguarda questi aspetti legati al rischio/beneficio e si raccomanda un monitoraggio molto stretto nella fase iniziale dopo il passaggio ad un altro trattamento.
Reazioni cutanee
In caso di stomatite ulcerativa, la somministrazione di leflunomide deve essere sospesa. Sono stati riportati casi molto rari di sindrome di Stevens–Johnson o di necrolisi epidermica tossica nei pazienti in terapia con leflunomide.
Appena si dovessero osservare reazioni della cute e/o delle mucose che destino il sospetto di reazioni così gravi, devono essere sospesi Arava ed altri trattamenti potenzialmente associati a tali reazioni e deve essere immediatamente iniziata una procedura di washout della leflunomide dall’organismo.
Un washout completo è essenziale in tali casi. La riesposizione a leflunomide è controindicata in tali casi (vedere paragrafo 4.3). Dopo l’uso di leflunomide sono stati segnalati psoriasi pustolosa e peggioramento della psoriasi. L’interruzione del trattamento può essere presa in considerazione in relazione alla malattia e all’anamnesi del paziente.
Infezioni
È noto che i medicinali immunosoppressivi – come leflunomide – possono predisporre i pazienti al pericolo di infezioni, incluse le infezioni opportunistiche. Possono manifestarsi infezioni più gravi in natura e per tale motivo possono richiedere un trattamento precoce e aggressivo.
Nel caso in cui insorga una infezione grave e incontrollata, può rendersi necessaria l’interruzione del trattamento con leflunomide e l’attuazione di una procedura di eliminazione accelerata del prodotto come descritto sotto. Sono stati riportati rari casi di Leucoencefalopatia Multiforme Progressiva (PML) in pazienti che assumono leflunomide in concomitanza ad altri immunosoppressori. Si deve prendere in considerazione il rischio di tubercolosi. Per quei pazienti con altri fattori di rischio per la tubercolosi si deve eseguire il test alla tubercolina.
Reazioni respiratorie
Durante il trattamento con leflunomide sono stati riportati casi di malattia interstiziale polmonare (vedere paragrafo 4.8). Il rischio che ciò di verifichi è maggiore in pazienti con anamnesi di malattia interstiziale polmonare. La malattia interstiziale polmonare è una patologia potenzialmente fatale che può manifestarsi in maniera acuta durante la terapia. I sintomi polmonari come tosse e dispnea possono essere una ragione per interompere la terapia e per ulteriori indagini.
Neuropatia periferica
In pazienti che ricevono Arava sono stati segnalati casi di neuropatia periferica. La maggior parte dei pazienti è migliorata dopo l’interruzione di Arava. Tuttavia c’è stata un’ampia variabilità nel decorso clinico, cioè in alcuni pazienti la neuropatia si è risolta e alcuni pazienti hanno avuto sintomi persistenti.
Età superiore ai 60 anni, farmaci neurotossici concomitanti e diabete possono aumentare il rischio di neuropatia periferica. Se un paziente che riceve Arava sviluppa neuropatia periferica, considerare l’interruzione della terapia di Arava ed effettuare la procedura di eliminazione del farmaco (vedere paragrafo 4.4).
Pressione arteriosa
La pressione arteriosa deve essere controllata prima dell’inizio della terapia con leflunomide e quindi periodicamente.
Procreazione (raccomandazioni per gli uomini) I pazienti di sesso maschile devono essere informati della possibile tossicità fetale maschio–mediata.
Durante il trattamento con leflunomide deve essere garantita anche una contraccezione affidabile. Non ci sono dati specifici sul rischio di tossicità fetale maschio–mediata. Comunque, non sono state effettuate sperimentazioni animali finalizzate alla valutazione di questo specifico rischio.
Per ridurre al minimo qualsiasi possibilità di rischio, il paziente che intende generare deve sospendere l’assunzione di leflunomide e, al contempo, assumere 8 g di colestiramina 3 volte al giorno per 11 giorni oppure 50 g di carbone attivo in polvere 4 volte al giorno per 11 giorni. Successivamente, in entrambi i casi, la concentrazione plasmatica di A771726 viene misurata una prima volta.
Quindi, la concentrazione plasmatica di A771726 deve essere di nuovo determinata dopo un intervallo di almeno 14 giorni. Se entrambe le concentrazioni plasmatiche sono inferiori a 0,02 mg/l e dopo un ulteriore periodo di attesa di almeno 3 mesi, il rischio di tossicità fetale è molto basso.
Procedura di washout
Si devono somministrare 8 g di colestiramina 3 volte al giorno. In alternativa, si devono somministrare 50 g di carbone attivo in polvere 4 volte al giorno. La durata di un washout completo è solitamente di 11 giorni. La durata può subire variazioni a seconda delle variabili cliniche o di laboratorio. Lattosio
Arava contine lattosio. I pazienti con rari problemi ereditari di intolleranza al galattosio, carenza di Lapp lattasi o malassorbimento di glucosio–galattosio, non devono assumere questo farmaco.
04.5 Interazioni
Sono stati effettuati studi di interazione solo negli adulti.
In caso di recente o contemporaneo uso di farmaci epatotossici o ematotossici o quando il trattamento con leflunomide è seguito dal trattamento con tali farmaci senza un periodo di washout (vedere anche la condotta da seguire per l’associazione con altri trattamenti, vedere paragrafo 4.4), può aumentare la frequenza di effetti indesiderati.
Pertanto, si raccomanda un più stretto monitoraggio degli enzimi epatici e dei parametri ematologici nella fase iniziale dopo il passaggio ad un altro trattamento. In uno studio effettuato su un numero ridotto di pazienti (n=30), nel corso del quale la somministrazione di leflunomide (10–20 mg/giorno) è stata associata a quella di metotrexato (10–25 mg/settimana), la concentrazione degli enzimi epatici è risultata aumentata di 2–3 volte in 5 pazienti su 30. In tutti i casi questi aumenti sono regrediti continuando l’assunzione di entrambi i farmaci (2 casi) o sospendendo la somministrazione della leflunomide (3 casi).
In altri 5 pazienti è stato osservato un aumento di più di 3 volte: tali aumenti regredivano continuando l’assunzione di entrambi i farmaci (2 casi) o sospendendo la somministrazione della leflunomide (3 casi). Nei pazienti con artrite reumatoide non è stata osservata alcuna interazione farmacocinetica fra la leflunomide (10–20 mg/die) ed il metotrexato (10–25 mg/settimana).
Si raccomanda che i pazienti che ricevono leflunomide non siano trattati con colestiramina o con carbone attivo in polvere, in quanto questo comporta una diminuzione rapida e significativa della concentrazione plasmatica di A771726 (il metabolita attivo della leflunomide; vedere anche paragrafo 5).
Si ritiene che il meccanismo responsabile di questo comportamento sia da ricercarsi nell’interruzione del ricircolo enteroepatico e/o nella dialisi gastrointestinale di A771726. La precedente somministrazione di farmaci antiinfiammatori non steroidei (FANS) e/o di corticosteroidi può essere continuata anche dopo aver avviato un trattamento con leflunomide. Non sono ancora esattamente conosciuti gli enzimi coinvolti nel processo di metabolizzazione della leflunomide e dei suoi metaboliti. Uno studio in vivo sull’eventuale interazione con la cimetidina (sostanza che inibisce in modo non specifico il citocromo P450) ha dimostrato assenza di interazione significativa.
Dopo somministrazione concomitante di una dose singola di leflunomide a soggetti che ricevevano dosi multiple di rifampicina (induttore non specifico del citocromo P450) sono stati osservati aumenti delle concentrazioni di picco di A771726 approssimativamente del 40%, senza significative modificazioni dell’area sotto la curva (AUC). Non è ancora noto il meccanismo che determina un tale effetto. Studi in vitro indicano che A771726 inibisce l’attività del citocromo P4502C9 (CYP2C9).
Nel corso di sperimentazioni cliniche non è emerso alcun problema di sicurezza in caso di somministrazione associata di leflunomide e di FANS metabolizzati da CYP2C9. È consigliabile prudenza in caso di somministrazione di leflunomide associata a farmaci diversi dai FANS, metabolizzati da CYP2C9, come la fenitoina, la warfarina, il fenprocumone e la tolbutamide.
In uno studio condotto su volontarie sane, che prevedeva la somministrazione concomitante di leflunomide e di un contraccettivo trifasico per uso orale contenente 30 mcg di etinilestradiolo, non è stata osservata riduzione di sorta della attività contraccettiva del suddetto farmaco; i parametri farmacocinetici di A771726 si sono attestati entro i valori previsti.
Vaccinazioni
Non sono disponibili dati clinici sull’efficacia e la sicurezza delle vaccinazioni durante trattamento con leflunomide. Tuttavia, la vaccinazione con vaccini vivi attenuati non è raccomandata. Per la somministrazione di un vaccino vivo attenuato, anche se successivamente alla sospensione del trattamento con Arava, si deve tenere conto della prolungata emivita della leflunomide.
04.6 Gravidanza e allattamento
Gravidanza Il metabolita attivo di leflunomide, A771726, si ritiene possa causare gravi anomalie congenite se somministrato durante lagravidanza. Arava è controindicato in gravidanza (vedere paragrafo 4.3).
Le donne in età fertile devono fare uso di un contraccettivo efficace durante e fino a 2 anni dopo il trattamento (vedere sotto “Periodo di attesa”) o fino a 11 giorni dopo il trattamento (vedere sotto “periodo di washout” abbreviato).
La paziente deve essere informata che, in presenza di qualsiasi ritardo del flusso mestruale o di qualsiasi altra ragione che faccia sospettare una gravidanza in atto, deve immediatamente informarne il medico che provvederà a prescriverle un test di gravidanza. Se questo risulta positivo, il medico e la paziente dovranno discutere i rischi eventualmente connessi con questa situazione.
È possibile che la rapida riduzione della concentrazione di metabolita attivo nel sangue (attuando la procedura di eliminazione del farmaco descritta più oltre), realizzata al primo ritardo del flusso mestruale, possa diminuire i rischi per il feto derivanti dalla leflunomide. In un piccolo studio prospettico in donne (n=64) divenute inavvertitamente gravide durante il trattamento con leflunomide, assunto per non più di tre settimane dopo il concepimento e che attuarono la procedura di eliminazione del farmaco, non sono state osservate differenze significative (p=0,13) nel tasso globale di difetti strutturali maggiori (5,4%) rispetto a entrambi i gruppi di confronto (4,2% nel gruppo con la malattia [n=108] e 4,2% nelle volontarie sane [n=78]).
In caso di donne trattate con leflunomide e che desiderano intraprendere una gravidanza, si raccomanda una delle seguenti procedure al fine di assicurare che il feto non sia esposto a concentrazioni tossiche di A771726 (concentrazione di riferimento inferiore a 0,02 mg/l).
Periodo di attesa
I livelli plasmatici di A771726 possono rimanere superiori a 0,02 mg/l per un periodo prolungato. La concentrazione può diminuire al di sotto di 0,02 mg/l dopo circa 2 anni dall’interruzione del trattamento con leflunomide. Dopo un periodo di attesa di 2 anni, la concentrazione plasmatica di A771726 viene misurata una prima volta. Quindi, la concentrazione plasmatica di A771726 deve essere determinata ancora dopo un intervallo di almeno 14 giorni.
Nessun rischio teratogeno è prevedibile se entrambe le concentrazioni plasmatiche sono inferiori a 0,02 mg/l. Per ulteriori informazioni sui prelievi da analizzare, per favore contattare il titolare dell’Autorizzazione all’Immissione in Commercio o il suo rappresentante locale (vedere paragrafo 7).
Procedura di washout
Dopo l’interruzione del trattamento con leflunomide: • devono essere somministrati 8 g di colestiramina 3 volte al giorno per un periodo di 11 giorni, • in alternativa, devono essere somministrati 50 g di carbone attivo in polvere 4 volte al giorno per un periodo di 11 giorni. Tuttavia, a seguito di entrambe le procedure di washout, è richiesta una verifica mediante 2 test separati da un intervallo di almeno 14 giorni ed un periodo di attesa di un mese e mezzo tra la prima volta che si ottiene una concentrazione plasmatica inferiore a 0,02 mg/l e la fecondazione.
Le donne potenzialmente fertili devono essere informate che è richiesto un periodo di attesa di 2 anni dopo l’interruzione del trattamento, prima di decidere una gravidanza. Se non si considera possibile un periodo di attesa di circa 2 anni con attuazione di forme affidabili di contraccezione, si potrà raccomandare l’adozione della procedura di washout. Sia la colestiramina che il carbone attivo in polvere possono influenzare l’assorbimento degli estrogeni e dei progestinici in modo tale che una contraccezione affidabile con contraccettivi orali potrebbe non essere garantita durante la procedura di washout con colestiramina o carbone attivo in polvere. Si raccomanda l’uso di metodi alternativi di contraccezione.
Allattamento
Studi condotti nell’animale indicano che la leflunomide o i suoi metaboliti passano nel latte materno. Le donne che allattano non devono pertanto assumere leflunomide.
04.7 Effetti sulla capacità di guidare veicoli e sull’uso di macchine
In caso di effetti indesiderati come capogiri, la capacità del paziente a concentrarsi ed a reagire prontamente può risultare alterata. In questi casi i pazienti devono astenersi dal guidare automobili e dall’usare macchinari.
04.8 Effetti indesiderati
Riassunto del profilo di sicurezza Di norma, gli effetti indesiderati più frequentemente riportati (≥1/100 – <1/10) con leflunomide sono: modesto aumento della pressione arteriosa, leucopenia, parestesia, cefalea, capogiri, diarrea, nausea, vomito, alterazioni della mucosa orale (ad esempio stomatite aftosa, ulcerazioni della bocca), dolore addominale, incremento della perdita dei capelli, eczema, rash (incluso rash maculopapulare), prurito, pelle secca, tenosinovite, incremento dei valori di CPK, anoressia, perdita di peso (generalmente non significativa), astenia, reazioni allergiche lievi ed aumento degli enzimi epatici (transaminasi (specialmente le ALT), meno spesso gamma–GT, fosfatasi alcalina, bilirubina).
Classificazione dei valori di frequenza attesi:
Molto comune (≥1/10); comune (≥1/100, <1/10); non comune (≥1/1.000, <1/100); raro (≥1/10.000, <1/1.000); molto raro (<1/10.000), non nota (la frequenza non puo essere definita sulla base dei dati disponibili). All’interno di ciascuna classe di frequenza, gli effetti indesiderati sono riportati in ordine decrescente di gravità.
Infezioni e infestazioniRaro: infezioni gravi, inclusa la sepsi che può essere fatale.
Come altri agenti potenziali immunosoppressori, leflunomide può aumentare la predisposizione alle infezioni, comprese le infezioni opportunistiche (vedere anche paragrafo 4.4).
Pertanto, l’incidenza globale delle infezioni può aumentare (in particolare di riniti, bronchiti e polmoniti). Tumori benigni, maligni e non specificati (cisti e polipi compresi) L’uso di alcuni agenti immunosoppressori aumenta il rischio di sviluppo di tumori maligni, specialmente di tipo linfoproliferativo.
Patologie del sistema emolinfopoietico Comune:
leucopenia (leucociti > 2 G/l) Non comune: anemia, lieve trombocitopenia (piastrine < 100 G/l) Raro: pancitopenia (probabilmente per un meccanismo antiproliferativo), leucopenia (leucociti < 2 G/l), eosinofilia Molto raro: agranulocitosi Un recente, concomitante o consecutivo uso di farmaci potenzialmente mielotossici può essere associato ad un rischio più elevato di effetti ematologici
Disturbi del sistema immunitario Comune: reazioni allergiche lievi Molto raro: reazioni anafilattiche/anafilattoidi gravi, vasculite, compresa vasculite cutanea necrotizzante Disturbi del metabolismo e della nutrizione Comune: incremento dei valori di CPK Non comune: ipopotassiemia, iperlipidemia, ipofosfatemia Raro: incremento dei valori di LDH Non nota: ipouricemia
Disturbi psichiatrici Non comune: ansia Patologie del sistema nervoso Comune: parestesia, cefalea, capogiri, neuropatia periferica Patologie cardiache Comune: modesto aumento della pressione arteriosa Raro: aumento grave della pressione arteriosa Patologie respiratorie, toraciche e mediastiniche Raro: malattia polmonare interstiziale (inclusa la polmonite interstiziale) che può essere fatale Patologie gastrointestinali Comune: diarrea, nausea, vomito, alterazioni della mucosa orale (ad esempio stomatite aftosa, ulcerazioni della bocca), dolore addominale. Non comune: disturbi del gusto Molto raro: pancreatite Patologie epatobiliari Comune: aumento degli indici di funzionalità epatica (transaminasi [specialmente ALT], meno spesso gamma–GT, fosfatasi alcalina, bilirubina) Raro: epatite, ittero/colestasi Molto raro: gravi danni epatici come insufficienza epatica e necrosi epatica acuta che possono essere fatali Patologie della cute e del tessuto sottocutaneo Comune: incremento della perdita dei capelli, eczema, rash (incluso rash maculopapulare), prurito, pelle secca Non comune: orticaria Molto raro: necrolisi epidermica tossica, sindrome di Stevens–Johnson, eritema multiforme Non nota: Lupus eritematoso cutaneo, psoriasi pustolosa o peggioramento della psoriasi Patologie del sistema muscoloscheletrico e del tessuto connettivo Comune: tenosinovite Non comune: rottura del tendine Patologie renali e urinarie Non nota: insufficienza renale Patologie dell’apparato riproduttivo e della mammella Non nota: riduzione marginale (reversibile) della concentrazione spermatica, della conta totale degli spermatozoi e della motilità progressiva rapida Patologie sistemiche e condizioni relative alla sede di somministrazione Comune: anoressia, perdita di peso (generalmente non significativa), astenia
04.9 Sovradosaggio
Sintomi Sono stati riportati casi di overdose cronica in pazienti che prendevano Arava a dosi giornaliere fino a cinque volte la dose giornaliera raccomandata e sono stati riportati casi di overdose acuta negli adulti e nei bambini. Non sono stati riportati eventi avversi nella maggior parte dei casi di overdose segnalati. Gli eventi avversi compatibili con il profilo di sicurezza di leflunomide sono stati: dolore addominale, nausea, diarrea, enzimi epatici elevati, anemia, leucopenia, prurito e rash.
Trattamento Nel caso di un sovradosaggio o tossicità, si raccomanda l’uso di colestiramina o carbone attivo per accelerare l’eliminazione del farmaco. La somministrazione orale di colestiramina a tre volontari sani alla dose di 8 g tre volte al giorno per 24 ore, ha diminuito i livelli plasmatici di A771726 di circa il 40% in 24 ore e dal 49% al 65% in 48 ore. È stato dimostrato che il carbone attivo (polvere in sospensione), somministrato per via orale o tramite sonda nasogastrica (50 g ogni 6 ore, per 24 ore), è in grado di ridurre le concentrazioni plasmatiche di A771726, il metabolita attivo della leflunomide, del 37% in 24 ore e del 48% in 48 ore. Se clinicamente necessario, queste procedure di washout possono essere ripetute. Studi sia con le emodialisi che con la CAPD (dialisi peritoneale ambulatoriale cronica) indicano che A771726, il metabolita primario di leflunomide, non è dializzabile.
05.0 PROPRIETÀ FARMACOLOGICHE
05.1 Proprietà farmacodinamiche Categoria farmacoterapeutica: sostanze ad azione immunosoppressiva selettiva, Codice ATC: L04AA13.
Farmacologia umana Leflunomide è un agente antireumatico in grado di modificare il decorso della malattia dotato di proprietà antiproliferative.
Farmacologia animale In modelli sperimentali di artrite reumatoide e di altre malattie autoimmuni e nei trapianti la leflunomide è attiva soprattutto se somministrata durante la fase di sensibilizzazione. La sostanza ha caratteristiche di immunomodulazione / immunosoppressione, ha azione antiproliferativa e presenta proprietà antiinfiammatorie.
La leflunomide mostra i suoi migliori effetti di protezione su modelli di animali con malattie autoimmuni quando somministrata allo stadio iniziale di progressione della malattia. In vivo, la leflunomide viene metabolizzata rapidamente e quasi completamente in A771726, che è attivo in vitro e si presume essere responsabile dell’effetto terapeutico.
Meccanismo di azione A771726, il metabolita attivo della leflunomide inibisce l’enzima diidroorotato deidrogenasi umano (DHODH) e mostra un’attività antiproliferativa.
Efficacia e sicurezza clinicaArtrite reumatoide
L’efficacia di Arava nel trattamento dell’artrite reumatoide è stata dimostrata in 4 sperimentazioni controllate (una di fase II e tre di fase III). Nella sperimentazione di fase II, studio YU203, 402 soggetti affetti da artrite reumatoide sono stati randomizzati al trattamento con placebo (n=102), leflunomide 5 mg/die (n=95), 10 mg/die (n=101) o 25 mg/die (n=104).
La durata del trattamento è stata di 6 mesi. Tutti i pazienti che hanno ricevuto leflunomide nelle sperimentazioni di fase III hanno assunto una dose iniziale di 100 mg per 3 giorni. Lo studio MN301 ha randomizzato 358 soggetti affetti da artrite reumatoide attiva al trattamento con leflunomide 20 mg /die (n=133), sulfasalazina 2 g/die (n=133) o placebo (n=92). La durata del trattamento è stata di 6 mesi.
Lo studio MN303 ha costituito una continuazione facoltativa in cieco per 6 mesi dello studio MN301 senza il gruppo placebo al fine di avere risultati comparativi a 12 mesi tra leflunomide e sulfasalazina.
Nello studio MN302, 999 soggetti affetti da artrite reumatoide attiva sono stati randomizzati al trattamento con leflunomide 20 mg/die (n=501) o metotrexato 7,5 mg/settimana, aumentato fino a 15 mg/settimana (n=498). L’aggiunta di folato era facoltativa e veniva utilizzata soltanto nel 10% dei pazienti.
La durata del trattamento è stata di 12 mesi.
Nello studio US301, 482 soggetti affetti da artrite reumatoide attiva sono stati randomizzati al trattamento con leflunomide 20 mg/die (n=182), metotrexato 7,5 mg/settimana, aumentato fino a 15 mg/settimana (n=182), o placebo (n=118). Tutti i pazienti hanno assunto folato 1 mg due volte al giorno. La durata del trattamento è stata di 12 mesi. La leflunomide ad una dose giornaliera di almeno 10 mg (da 10 a 25 mg nello studio YU203, 20 mg negli studi MN301 e US301) è risultata superiore in modo statisticamente significativo rispetto al placebo nel diminuire i segni ed i sintomi dell’artrite reumatoide in tutte e tre le sperimentazioni controllate vs placebo.
Le percentuali di risposta secondo l’ACR (American College of Rheumatology) nello studio YU203 sono state 27,7% per il placebo, 31,9% per 5 mg/die, 50,5% per 10 mg/die e 54,5% per 25 mg/die di leflunomide. Nelle sperimentazioni di fase III, le percentuali di risposta secondo l’ACR per leflunomide 20 mg/die vs placebo sono state di 54,6% vs 28,6% (studio MN301) e 49,4% vs 26,3% (studio US301).
Dopo 12 mesi di trattamento attivo, le percentuali di risposta secondo l’ACR nei pazienti trattati con leflunomide sono state di 52,3% (studi MN301/303), 50,5% (studio MN302) e 49,4% (studio US301), in confronto al 53,8% (studi MN301/303) nei pazienti trattati con sulfasalazina, e al 64,8% (studio MN302) e 43,9% (studio US301) nei pazienti trattati con metotrexato.
Nello studio MN302 la leflunomide è stata significativamente meno efficace del metotrexato. Tuttavia, nello studio US301 non è stata osservata alcuna differenza significativa tra la leflunomide ed il metotrexato nei parametri di efficacia primari. Nessuna differenza è stata osservata tra leflunomide e sulfasalazina (studio MN301).
L’effetto del trattamento con leflunomide è risultato evidente dopo 1 mese, si è stabilizzato fra 3 e 6 mesi e si è protratto nel corso del trattamento. Uno studio di non inferiorità, randomizzato, in doppio cieco, a gruppi paralleli, ha confrontato l’efficacia relativa di due diverse dosi giornaliere di mantenimento di leflunomide, 10 mg e 20 mg. Dagli esiti è possibile giungere alla conclusione che i risultati di efficacia della dose di mantenimento di 20 mg sono stati più favorevoli mentre, d’altro canto, i risultati di sicurezza sono più favorevoli alla dose di mantenimento di 10 mg.
Popolazione pediatrica
Leflunomide è stata studiata in uno studio multicentrico, controllato vs farmaco attivo, randomizzato in doppio–cieco, condotto su 94 pazienti (47 per braccio) affetti da artrite reumatoide giovanile a decorso poliarticolare. I pazienti avevano un’età compresa tra 3 e–17 anni con artrite reumatoide giovanile attiva a decorso poliarticolare, indipendentemente dal tipo di inizio e non erano stati trattati in precedenza con metotrexato o leflunomide.
In questo studio, la dose di carico e di mantenimento di leflunomide è stata calcolate in base a tre categorie di peso: <20 kg, 20–40 kg e >40 kg. Dopo 16 settimane di trattamento, la differenza nel tasso di risposte secondo la Definizione del miglioramento per l’artite reumatoide giovanile (DOI ≥30 %) è risultata statisticamente significativa (p=0,02) per il gruppo trattato con metotrexato. Nei pazienti che hanno risposto, tale risposta si è mantenuta per 48 settimane (vedere paragrafo 4.2).
Il profilo di effetti indesiderati è apparso simile con leflunomide e con metotrexato; tuttavia la dose utilizzata nei pazienti a più basso peso ha comportato un’esposizione relativamente bassa (vedere paragrafo 5.2). Tali dati non permettono di raccomandare una dose efficace e sicura.
Artrite psoriasica L’efficacia di Arava è stata dimostrata in uno studio (3L01) controllato, randomizzato, in doppio cieco, in 188 pazienti affetti da artrite psoriasica, trattati con 20 mg al giorno. La durata del trattamento è stata di 6 mesi. Leflunomide 20 mg al giorno è risultata significativamente superiore al placebo nel ridurre i sintomi dell’artrite nei pazienti con artrite psoriasica: il PsARC (criteri di risposta al trattamento dell’artrite psoriasica) ha messo in evidenza il 59% dei responder nel gruppo trattato con leflunomide nei confronti del 29,7% del gruppo trattato con placebo a 6 mesi (p < 0,0001). Gli effetti della leflunomide nel migliorare la funzionalità e nella riduzione delle lesioni cutanee sono risultati modesti.
Studi di post–marketing
Uno studio randomizzato ha valutato l’efficacia clinica del tasso di risposta in nuovi pazienti affetti da DMARD (n=121) con AR iniziale, che hanno ricevuto in doppio cieco in due gruppi paralleli o 20 mg o 100 mg di leflunomide durante i primi tre giorni di trattamento.
La fase iniziale è stata seguita da un periodo di mantenimento in aperto di tre mesi durante il quale entrambi i gruppi hanno ricevuto 20 mg di leflunomide al giorno. Non è stato osservato nessun aumento del beneficio complessivo nel gruppo di pazienti che ha ricevuto la terapia con la dose di carico.
I dati di sicurezza ottenuti da entrambi i gruppi in trattamento sono stati coerenti con il profilo noto di sicurezza della leflunomide, tuttavia, l’incidenza di effetti indesiderati gastrointestinali e di aumento degli enzimi epatici ha avuto la tendenza ad essere più alta nei pazienti che hanno ricevuto una dose di carico di 100 mg di leflunomide.
05.2 Proprietà farmacocinetiche
La leflunomide viene convertita rapidamente nel suo metabolita attivo, A771726, mediante metabolismo di primo passaggio (apertura dell’anello) che si realizza a livello della parete intestinale e del fegato. In uno studio condotto su tre volontari sani con leflunomide marcata con 14C non è stata rilevata presenza di leflunomide immodificata nel plasma, nelle urine e nelle feci. In altri studi, il riscontro di leflunomide non modificata nel plasma è stato raro e, comunque, con livelli nell’ordine di grandezza di ng/ml. Il solo metabolita radiomarcato presente nel plasma è stato A771726. Questo metabolita è responsabile essenzialmente di tutta l’attività di Arava in vivo.
Assorbimento
I dati di escrezione ottenuti dallo studio con 14C indicano un assorbimento non inferiore allo 82–95% della dose somministrata. Il tempo occorrente perché la concentrazione di A771726 nel plasma raggiunga valori di picco varia molto; i livelli di picco plasmatico possono essere riscontrati fra 1 e 24 ore dopo singola somministrazione.
La leflunomide può essere somministrata in concomitanza con l’assunzione di cibo dato che l’entità dell’assorbimento è simile tanto dopo assunzione di cibo che a digiuno. Data l’emivita molto protratta di A771726 (circa 2 settimane) nel corso di studi clinici è stata impiegata una dose di carico di 100 mg per 3 giorni, in modo da facilitare un rapido raggiungimento dello steady–state delle concentrazioni di A771726.
In assenza di una dose di carico, si stima che siano necessari quasi 2 mesi di somministrazione per raggiungere lo steady state delle concentrazioni plasmatiche. I risultati ottenuti in studi con somministrazione di dosi ripetute a pazienti affetti da artrite reumatoide hanno dimostrato che i parametri farmacocinetici di A771726 presentano un andamento lineare entro l’intervallo di dosi impiegate (5–25 mg).
In questi studi, l’effetto clinico era strettamente correlato con le concentrazioni plasmatiche di A771726 e con la dose giornaliera di leflunomide. Con dosi di 20 mg/die, la concentrazione media plasmatica di A771726 allo steady–state è di circa 35 mcg/ml. Allosteady–state le concentrazioni plasmatiche risultano pari a circa 33–35 volte quelle relative alla somministrazione di una singola dose.
Distribuzione
Nel plasma umano, A771726 è legato estesamente alle proteine (albumina). La frazione non legata di A771726 è circa lo 0,62%. Il legame di A771726 risulta lineare alle concentrazioni comprese nell’intervallo terapeutico.
Il legame è lievemente inferiore e maggiormente variabile nel plasma dei pazienti con artrite reumatoide o con insufficienza renale cronica.
L’esteso legame di A771726 alle proteine potrebbe causare lo spostamento di altri farmaci ad elevato legame proteico. Comunque, studi sull’interazione di legame con le proteine plasmatiche condotti in vitro impiegando concentrazioni di warfarina clinicamente significative non hanno dimostrato interazioni. Studi analoghi hanno dimostrato che ibuprofene e diclofenac non spiazzano A771726, mentre la frazione libera di A771726 va incontro ad un aumento di 2–3 volte in presenza di tolbutamide.
A771726 è in grado di spostare l’ibuprofene, il diclofenac e la tolbutamide, ma la frazione libera di questi farmaci è aumentata soltanto del 10–50%. Non vi sono indicazioni che questi effetti siano clinicamente rilevanti. Coerentemente con il suo accentuato legame proteico, A771726 presenta un basso volume di distribuzione apparente (circa 11 1itri). Non vi è captazione preferenziale da parte degli eritrociti.
Biotrasformazione
La metabolizzazione di leflunomide dà luogo alla formazione di un metabolita primario (A771726) e di numerosi metaboliti minori, incluso TFMA (4–trifluorometilalanina).
La biotrasformazione metabolica della leflunomide in A771726 e la successiva metabolizzazione di A771726 non sono controllate da un singolo enzima ed è stato dimostrato che esse si verificano nelle frazioni cellulari microsomiali e citosoliche.
Studi sulle interazioni, condotti con cimetidina (inibitore non specifico del citocromo P450) e rifampicina (induttore non specifico del citocromo P450), hanno evidenziato che, in vivo, gli enzimi CYP non sono coinvolti se non in misura ridotta nel metabolismo della leflunomide.
Eliminazione
L’eliminazione di A771726 ha luogo lentamente ed è caratterizzata da una clearance apparente di circa 31 ml/h. Nei pazienti, l’emivita di eliminazione è approssimativamente di 2 settimane.
Dopo somministrazione di una dose di leflunomide radiomarcata, la radioattività risulta escreta in pari misura attraverso le feci (probabilmente attraverso eliminazione biliare) e le urine. A771726 è stato riscontrato nelle feci e nelle urine anche a distanza di 36 giorni da una singola somministrazione.
I principali metaboliti urinari sono costituiti da prodotti glucuronidi derivati dalla leflunomide (presenti maggiormente nei campioni prelevati nelle prime 24 ore) e da un derivato dell’acido ossanilico di A771726. Il principale componente reperito nelle feci è lo A771726.
Nell’uomo si è osservato che la somministrazione per os di una sospensione di polvere di carbone attivo o di colestiramina induce un rapido e significativo aumento della velocità di eliminazione di A771726 e del declino della concentrazione plasmatica (vedere paragrafo 4.9). Si pensa che questo sia dovuto ad un meccanismo di dialisi gastrointestinale e/o all’interruzione del ricircolo enteroepatico.
Insufficienza renale
La leflunomide è stata somministrata come dose singola orale (100 mg) a 3 pazienti emodializzati ed a 3 pazienti in dialisi peritoneale continua ambulatoriale (CAPD). La farmacocinetica di A771726 nei soggetti in CAPD è apparsa simile a quella dei volontari sani: Una più rapida eliminazione di A771726 è stata osservata nei soggetti in emodialisi, tale eliminazione non era causata dall’estrazione del farmaco nei liquidi di dialisi.
Insufficienza epatica
Non sono disponibili dati sul trattamento di pazienti affetti da insufficienza epatica. Il metabolita attivo, l’A771726, si lega fortemente alle proteine plasmatiche e viene eliminato mediante escrezione biliare previo metabolismo epatico; questi processi possono essere compromessi da una disfunzione epatica.
Popolazione pediatrica
La farmacocinetica di A771726 in seguito a somministrazione orale di leflunomide è stata valutata in 73 pazienti pediatrici con artrite reumatoide giovanile a decorso poliarticolare di età compresa tra 3 e 17 anni. I risultati di un’analisi farmacocinetica di popolazione di questi studi clinici hanno dimostrato che i pazienti pediatrici con peso corporeo £40 kg hanno un’esposizione sistemica a A771726 ridotta (valutata tramite Css) rispetto ai pazienti adulti con artrite reumatoide (vedere paragrafo 4.2).
Anziani
I dati farmacocinetici relativi ai pazienti anziani (> 65 anni) sono limitati ma mostrano una buona corrispondenza con quelli ottenuti in giovani adulti.
05.3 Dati preclinici di sicurezza
Studi di tossicità acuta sono stati condotti mediante somministrazione orale ed intraperitoneale di leflunomide nel topo e nel ratto. La somministrazione orale ripetuta di leflunomide a topi (fino a 3 mesi), ratti e cani (fino a 6 mesi) e scimmie (fino ad 1 mese) ha evidenziato che i principali organi bersaglio della tossicità sono il midollo spinale, il sangue, il tratto gastrointestinale, la cute, la milza, il timo ed i linfonodi.
Gli effetti principali (rappresentati da anemia, leucopenia, riduzione del numero delle piastrine e panmielopatia) riflettono il meccanismo d’azione di base del farmaco (inibizione della sintesi del DNA). Nel ratto e nel cane sono stati individuati corpuscoli di Heinz e/o corpuscoli di Howell–Jolly. Altri effetti, a carico di cuore, fegato, cornea e tratto respiratorio, possono essere interpretati come infezioni indotte da immunosoppressione. La tossicità negli animali è stata evidenziata con dosi equivalenti alle dosi terapeutiche umane.
La leflunomide non è mutagena. Tuttavia, il metabolita secondario TFMA (4‑trifluorometilalanina) ha indotto in vitroclastogenicità e mutazioni puntiformi. Attualmente, non sono disponibili sufficienti informazioni sulla sua capacità di espletare analogo effetto in vivo. In uno studio di cancerogenicità nel ratto, la leflunomide si è dimostrata priva di potenziale cancerogeno. In un analogo studio nel topo è stata riscontrata una maggiore frequenza di linfomi maligni nei maschi del gruppo a più elevato dosaggio: tale effetto è stato attribuito all’attività immunosoppressiva della leflunomide. Nel topo femmina è stata osservata un aumento dose–dipendente dell’incidenza di adenomi bronchiolo–alveolari e di carcinomi del polmone. La rilevanza dei risultati degli studi sui ratti nella pratica clinica di leflunomide è dubbia.
Leflunomide non ha presentato proprietà antigeniche nei modelli animali. Alle dosi proprie dell’ambito terapeutico umano, la leflunomide ha evidenziato proprietà embriotossiche e teratogene se somministrata a ratti e conigli. Inoltre, in studi di tossicità, la somministrazione ripetuta di leflunomide ha indotto effetti avversi a carico degli organi riproduttivi maschili. La fertilità non risultava ridotta.
fonte reperibile di seguitohttp://www.torrinomedica.it/farmaci/schedetecniche/Arava_20_Mg.asp#axzz35V2Y581b
Cosa dichiara la FDA?
Annuncio Sicurezza
[07-13-2010]
La US Food and Drug Administration (FDA) sta aggiungendo informazioni su danni epatici gravi al Boxed Warning di Arava (leflunomide) – un farmaco usato per trattare l’artrite reumatoide – per evidenziare il rischio di una grave fegato lesioni nei pazienti che usano questo farmaco e come tale rischio può essere ridotto. FDA precedentemente richiesto un boxed warning che indica che la leflunomide è stato controindicato nelle donne in gravidanza, o donne in età fertile che non stavano usando la contraccezione affidabile.
Anche se un avvertimento in grassetto sul grave danno epatico è stato aggiunto all’etichetta farmaco leflunomide nel 2003, la FDA ha stabilito che le informazioni sui gravi danni epatici dovrebbe essere incluso nel Boxed Warning per evidenziare l’importanza della selezione appropriata paziente prima di iniziare il trattamento e il monitoraggio al termine del trattamento ha iniziato.
La decisione di aggiungere informazioni sui gravi danni epatici al Boxed Warning è basata su di FDA 2010 Recensione di segnalazioni di eventi avversi, che ha individuato 49 casi di grave danno epatico, tra cui 14 casi di insufficienza epatica fatale, tra l’agosto 2002 e il maggio 2009. In questa recensione , il maggior rischio di danno epatico è stato osservato nei pazienti che assumono altri farmaci noti per causare danno epatico, e pazienti con malattia epatica pre-esistente .
Gli operatori sanitari devono essere consapevoli del rischio di danno epatico grave con questo farmaco, e di garantire adeguata selezione dei pazienti e il monitoraggio …